
Mais de 20 portadores de um tipo de cegueira causada por um defeito genético e conhecida como amaurose congênita de Leber (ACL) voltaram a enxergar graças a um tratamento desenvolvido por pesquisadores da University of Pennsylvania e do The Children’s Hospital of Philadelphia – ambos nos Estados Unidos.
Um dos líderes do grupo é o brasileiro Valder Arruda, que apresentou resultados dos testes já realizados durante o evento “Advanced Topics in Genomics and Cell Biology”, realizado entre os dias 4 e 6 de agosto na Universidade Estadual de Campinas (Unicamp) com apoio da FAPESP.
“Demonstramos a segurança e a eficácia do tratamento e estamos na fase de encontrar a dose capaz de ter efeito terapêutico em todos os pacientes. Acredito que essa possa ser a primeira terapia gênica aprovada para uso comercial pelo FDA [Food and Drug Administration, agência de vigilância sanitária americana]”, disse Arruda, ex-professor da Unicamp e atualmente docente da University of Pennsylvania. Até chegar a essa fase, foram muitos anos de estudo e ensaios.
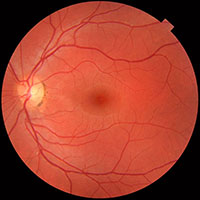
Em entrevista à Agência FAPESP, o pesquisador explicou que são conhecidos pelo menos 15 genes distintos que, mutados, resultam em cegueira congênita do tipo ACL. Todos eles causam uma degeneração progressiva da retina que tem início no nascimento.
“Desde muito cedo, as crianças apresentam visão prejudicada e movimentos oculares incomuns. Geralmente, ficam cegas durante a adolescência”, contou Arruda.
A terapia desenvolvida pelo grupo da University of Pennsylvania, que também conta com a liderança da oftalmologista Jean Bennett, visa tratar portadores de uma mutação no gene que codifica a proteína RPE65, envolvida no metabolismo da vitamina A – um nutriente essencial para a saúde dos tecidos oculares.
“Optamos por começar as pesquisas com o gene RPE65 porque existem alguns cachorros com a mesma mutação espontânea encontrada em humanos, o que facilita a realização dos testes pré-clínicos. Mas a tecnologia pode ser adaptada para outras formas de amaurose congênita e há grupos em vários países realizando pesquisas com esse objetivo. Claro que cada caso terá suas particularidades”, disse Arruda.
O método consiste em inserir uma cópia sadia do gene RPE65 dentro de um vírus modificado. O vetor escolhido pelos pesquisadores foi o vírus adeno-associado (AAV, na sigla em inglês), uma espécie do gênero parvovírus que não causa doenças em humanos.
“Retiramos o material genético do AAV e inserimos em seu lugar uma cópia do RPE65 e um promotor – molécula que vai mandar uma mensagem para ativar a expressão do gene. A ideia é aproveitar a capacidade natural do vírus de infectar as células humanas e depositar nelas seu material genético. Dessa forma, as células da retina passam a expressar a proteína RPE65 que antes era deficiente”, explicou o pesquisador.
Em 2001, o grupo de Bennett publicou na revista Nature Genetics resultados promissores dos testes feitos em cães e, em seguida, com a parceria de Arruda, foram iniciados os primeiros ensaios em humanos.
“No ensaio clínico de fase 1, o objetivo era atestar a segurança do tratamento. Injetamos uma dose baixa do vetor viral em três adultos portadores da mutação. Normalmente, ensaios de fase 1 são feitos apenas em pacientes sadios, mas no caso da terapia gênica é diferente”, explicou Arruda.
Os primeiros exames feitos após o tratamento revelaram que a pupila dos voluntários, que antes não reagia à variação de luminosidade, passou a responder quase da mesma forma que a de uma pessoa com visão normal.
“Três semanas após a aplicação do vetor viral, uma das pacientes disse que estava conseguindo ver o contorno dos móveis de seu apartamento. A princípio, achei que fosse apenas efeito placebo, mas, poucas semanas depois, outro paciente relatou algo parecido”, contou Arruda.
Testes posteriores feitos por uma equipe independente comprovaram que os três voluntários haviam recuperado a visão parcialmente, contrariando o conceito predominante na área de neurociência de que o cérebro de uma pessoa que passou muitos anos sem enxergar teria perdido para sempre a capacidade de interpretar os sinais captados pela retina.
“Essa primeira paciente italiana tinha 36 anos. Por meio de exames de ressonância magnética funcional, mostramos que as áreas do córtex visual que estavam inativas antes do tratamento passaram a apresentar atividade 30 dias após a injeção do vetor viral. E essa atividade estava bem maior 90 dias depois. Esses resultados podem potencialmente questionar o grande dogma da neurociência”, disse Arruda.
Os primeiros ensaios feitos com 12 pacientes, entre ele um menino de 8 anos, foram divulgados em 2009 na revista The Lancet e mostraram que a eficácia do tratamento estava diretamente relacionada com seu início precoce.
“Normalmente, são permitidos apenas maiores de 18 anos nos ensaios de fase 1. Mas, como se trata de uma doença degenerativa, pode não restar nenhum tecido retiniano para ser restaurado se demorarmos muito para começar o tratamento. Por conta disso, tivemos autorização para incluir crianças. Agora, estamos iniciando os ensaios de fase 3 e baixamos a idade mínima para 4 anos”, contou Arruda.
Até o momento, 24 pacientes de países como Itália, Holanda, Bélgica, Estados Unidos e Austrália já receberam a injeção intraocular com o vetor viral – primeiro em um olho e, posteriormente, no outro – e apresentaram melhora visual sem qualquer evidência de toxicidade ou reação imunológica.
“Temos uma menina de 9 anos que passou a brincar de esconde-esconde, um menino de 16 anos que conseguiu tirar carteira de motorista e muitos adultos que se tornaram independentes. Todos se sentem mais confiantes”, comemorou Arruda.
Hemofilia
Antes de se unir à equipe de Bennett no tratamento de doenças da retina, Arruda – hematologista de formação – já vinha testando uma metodologia semelhante no tratamento de uma das formas de hemofilia, doença genética caracterizada por problemas de coagulação sanguínea.
“Existem dois tipos de hemofilia: A e B. A primeira ocorre por deficiência de uma proteína conhecida como fator VIII de coagulação do sangue e, a hemofilia B, por deficiência do fator IX. São necessários níveis adequados de ambas as proteínas no plasma para que a cascata de coagulação funcione corretamente quando necessário. Portanto, quando qualquer uma delas está deficiente, a manifestação é a mesma”, explicou Arruda.
A doença é considerada leve e pode passar despercebida por muitos anos quando os níveis do fator de coagulação deficiente – seja o VIII ou o IX – no plasma estão acima de 5% da quantidade encontrada no sangue de pessoas não hemofílicas. Quando o nível está entre 1% e 5%, a doença é considerada moderada. Abaixo de 1%, é considerada grave. Nesses casos, sangramentos ocorrem de forma espontânea, mesmo sem um trauma importante.
“Quanto maior a gravidade, mais frequentes são complicações como sangramento nas articulações, nos músculos ou intracraniano. O paciente grave costuma sangrar uma vez por mês. O moderado, duas ou três vezes por semestre. O leve pode passar um ano inteiro sem sangrar”, disse Arruda.
O tratamento hoje disponível, explicou o pesquisador, é a reposição do fator de coagulação deficiente, mas a proteína permanece por um curto período no sangue e precisa novamente ser reposta. A terapia pode ser feita de forma preventiva rotineiramente nos casos mais severos; porém, a grande limitação é o custo, que pode ultrapassar US$ 200 mil por ano para uma criança entre 10 e 15 anos de idade.
“No Brasil, o Sistema Único de Saúde (SUS), na grande maioria dos casos, oferece o fator de coagulação apenas quando o paciente já apresenta sangramento ou caso vá passar por um procedimento cirúrgico, por exemplo. Mas, cada vez que ocorre um sangramento, a região fica mais comprometida e sangra mais facilmente da próxima vez. O ideal é o tratamento profilático”, disse Arruda.
Com o objetivo de obter um método curativo, os pesquisadores da University of Pennsylvania começaram há mais de 15 anos a desenvolver um AAV modificado com uma cópia do gene responsável pela produção do fator IX. Os testes e ensaios passaram por diversas fases.
“Embora a hemofilia B não seja a forma mais prevalente, começamos por esse gene porque ele é muito menor que o gene do fator VIII e seria mais fácil para inseri-lo no vetor viral. Agora, estamos desenvolvendo uma versão da terapia para a hemofilia A. O objetivo é manter continuamente os níveis do fator de coagulação sanguínea acima de 1% do normal, para diminuir a necessidade do tratamento de reposição. Se conseguirmos manter acima de 6%, o paciente só precisaria repor a proteína caso fosse submetido a uma cirurgia”, contou Arruda.
Os primeiros testes feitos com uma linhagem de cachorros portadora de hemofilia do tipo B grave foram publicados em 2002 na revista Blood. Os animais foram seguidos por dois anos e, após uma única injeção, mantiveram estáveis os níveis plasmáticos de fator IX entre 6% e 9%. Até hoje são considerados portadores de hemofilia leve.
Na mesma época, teve início o primeiro ensaio clínico com sete voluntários adultos e, pela primeira vez, um vetor viral foi infundido no fígado humano pela artéria hepática, por meio de um cateter.
Embora a abordagem tenha se mostrado segura, para surpresa dos pesquisadores, os níveis plasmáticos de fator IX, que haviam aumentado logo depois da aplicação do tratamento, começaram a cair após algumas semanas.
Novos estudos revelaram que as células do fígado, que haviam começado a produzir o fator IX graças à terapia gênica, estavam sendo destruídas pelo sistema imunológico, que reconheceu a proteína do vetor viral como um corpo estranho. Tal efeito não havia sido observado nos cães e, posteriormente, não foi visto em macacos.
“Possivelmente, esses pacientes já haviam sido infectados pelo AAV anteriormente e haviam criado anticorpos contra a proteína do envelope do vírus”, disse Arruda.
Os resultados desse primeiro teste clínico foram divulgados em 2006 na revista Nature Medicine.
Em parceria com outros colaboradores do Childrens Hospital of Philadelphia, um grupo da St. Jude Children’s Research Hospital, também nos Estados Unidos, realizou um novo ensaio clínico de fase 1. Desta vez, os pesquisadores acompanharam de perto as alterações nas enzimas hepáticas e nos níveis de fator IX do sangue.
Ao menor sinal de que os hepatócitos estavam sendo destruídos pelo sistema imunológico, era administrada uma droga imunossupressora. A estratégia funcionou e os resultados foram divulgados em 2011 no The New England Journal of Medicine.
“A droga imunossupressora foi usada apenas durante oito semanas, pois com o tempo a proteína do envelope viral é degradada e o sistema imunológico para de atacar os hepatócitos. Um dos pacientes manteve entre 6% e 8% do normal os níveis de fator IX no plasma, enquanto outro que demorou mais tempo para receber a imunossupressão caiu para 3%. É preciso agir rapidamente”, disse Arruda.
Mesmo nos casos em que os níveis ficaram entre 1% e 2% do normal, acrescentou o pesquisador, já foi possível reduzir em 77% a necessidade de reposição da proteína.
Aprimoramento do vetor viral
Atualmente, há três grupos de pesquisa americanos testando metodologias para aperfeiçoar o vetor viral usado no tratamento da hemofilia B. No St. Jude Children’s Research Hospital foi inserido o gene do fator IX em um outro sorotipo do AAV que tem maior tendência de infectar o fígado. Dessa forma, a aplicação da terapia não precisa mais ser feita por meio de cateter diretamente no órgão alvo – basta uma injeção endovenosa simples.
Já o grupo de Arruda faz testes com uma variante da proteína do fator IX descrita em 2009 no The New England Journal of Medicine.
“Descobrimos em um morador da cidade de Pádua, na Itália, outra mutação que, ao contrário da hemofilia, aumenta a capacidade de coagulação sanguínea e predispõe os portadores a sofrer de trombose. Esse paciente tem os mesmos níveis de fator IX que uma pessoa saudável, mas uma alteração na sequência peptídica torna a proteína oito vezes mais eficaz para coagular o sangue”, explicou.
Testes com um vetor viral contendo a proteína do fator IX com a sequência peptídica alterada para mimetizar a mutação encontrada em Pádua foram bem-sucedidos em cães e o grupo da University of Pennsylvania inicia agora os primeiros ensaios de fase 1 em humanos.
“Com uma proteína oito vezes mais eficiente na coagulação sanguínea, podemos usar uma dose bem menor do vetor viral no tratamento da hemofilia B, o que diminui o risco de toxicidade hepática”, disse o pesquisador.
Quando ainda estava no Brasil, Arruda coordenou e participou de projetos de pesquisa apoiados pela FAPESP. Arruda mantém colaborações com pesquisadores da Unicamp, entre os quais Margareth Ozelo, diretora da Divisão de Hematologia do Departamento de Clínica Médica da Unicamp – o que permitiu a inclusão de um paciente brasileiro nos ensaios clínicos da terapia contra hemofilia.
“Nesses últimos anos, mais de dez estudantes brasileiros fizeram estágio comigo na Filadélfia e hoje estão aplicando em pesquisas na Unicamp a metodologia do AAV”, contou Arruda.
